人工智能和生物分子预测软件时代的神经精神药理学
快速阅读: 《Nature.com》消息,1865年提出SAR概念,后发展为QSAR,结合机器学习推动药物发现。AI助力高精度构建蛋白质三维结构,提升对生物分子活动的理解。
1865年,克鲁姆-布朗和弗雷泽提出了结构-活性关系(SAR)的概念[1]。SAR描述了化学结构与其预测的生物活性之间的关系,为科学家提供了一个框架,用于研究其功效和效力。通过实验性地改变这些要素,我们可以设计出能够利用或避免特定治疗结果的分子(即减少细胞毒性或增加活性)。该理论的现代应用将化学分子与其生物活性之间的数学关系结合进来,称为定量结构-活性关系(QSAR)[2]。随着时间的推移,QSAR发展成了基于机器学习(ML)的技术,能够提取大量数据集,从而成为药物发现的关键组成部分[3]。这些生物分子的定量进步在多个领域持续发展,并扩展了当今的生物技术领域。由于这些突破,我们现在能够应用这些技术来研究蛋白质和其他生物分子,改变了我们对神经精神药理学的研究方法。
氨基酸链可以分为多种类别,每种类别有助于定义分子的结构组件或生理功能。由此产生的蛋白质具有复杂的构象,可能有多种功能形式,每种形式都可能与其他蛋白质或信号分子相互作用[4]。这些相互作用的精确性质主要由蛋白质的折叠和展开所决定,因此代表了生物体内生物分子活动的关键机制[5]。然而,尽管蛋白质折叠本身被认为对细胞信号传导至关重要,但我们在建模和预测这些过程,以及开发能够模拟这些过程的新化合物方面,面临更大的挑战。
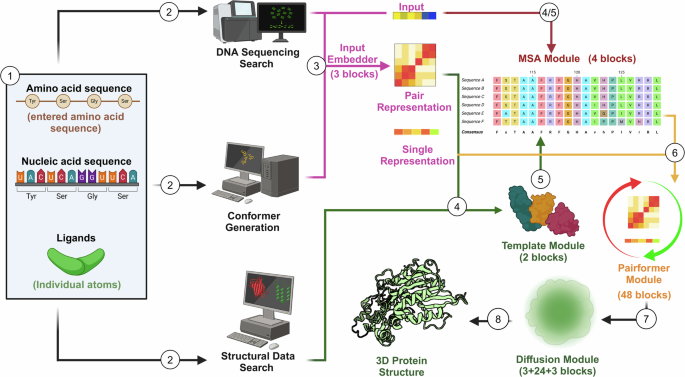
从结构角度来看,人工智能(AI)的使用使我们能够在高精度下快速构建三维蛋白质结构[6,7]。这项工作自最初尝试用橡皮泥模型可视化蛋白质分子以来取得了显著进展。虽然橡皮泥模型能够实现折叠的可视化,但其用途仅限于结构较为简单的蛋白质。X射线晶体学和电子显微镜使这种可视化达到了更加真实的图像,这些模式是通过计算机计算得出的[8]。尽管对于理解孤立的蛋白质很有用,但它仍然无法解决蛋白质如何变化的问题。这就是为什么在上个世纪花了数十年时间才生成蛋白质模型;因为没有方法可以可视化氨基酸在众多三级结构或形状中如何结合并折叠[9]。
(以上内容均由Ai生成)







